
量子アニーラを用いたドッキング計算 (2023/04-)
量子アニーラ (quantum annealer) とは、量子コンピュータの一種であり、特定の形式の組合せ最適化問題の局所最適解を高速に探索できると期待されている計算機です。そこで、この量子アニーラをタンパク質-リガンドのドッキング計算(タンパク質の立体構造に基づき候補化合物の結合様式と結合親和性を予測する手法)へ応用することを目指しています。そのために、ドッキング計算を量子アニーラで解ける形式に変換し、QUBO(quadratic unconstrained binary optimization)問題として定式化しました。具体的には、リガンド分子を複数のフラグメント(部分構造)に分割し、それぞれの配置をバイナリ変数で表現することで、ドッキングの探索空間をQUBOとして表現しています。
- Keisuke Yanagisawa, Takuya Fujie, Kazuki Takabatake, Yutaka Akiyama. “QUBO Problem Formulation of Fragment-Based Protein–Ligand Flexible Docking”, Entropy, 26(5): 397, 2024/4. DOI: 10.3390/e26050397
共溶媒分子動力学法 (2019/04-)
共溶媒分子動力学 (mixed-solvent molecular dynamics; MSMD) 法は、 タンパク質を溶質、水分子と共溶媒分子を溶媒とした分子動力学 (molecular dynamics; MD) 法です。 共溶媒分子がタンパク質表面のどこに、どの程度存在したか?という情報を基にホットスポット検出や結合親和性評価を行ったり 共溶媒分子によってタンパク質構造の変化が誘発されることを利用したcryptic結合部位(隠された結合部位)発見などに活用されています。
このMSMD法について、様々な提案を行ってきています。
共溶媒の相互作用プロファイルの設計 (Inverse MSMD)
通常の共溶媒MDでは、タンパク質表面のどの位置に共溶媒分子が存在しやすいか(存在確率分布)をマッピングします。これに対しinverse MSMDでは、共溶媒分子の周囲にどのような種類のタンパク質残基が現れやすいかという「共溶媒側から見た相互作用プロファイル」の推定を行います。
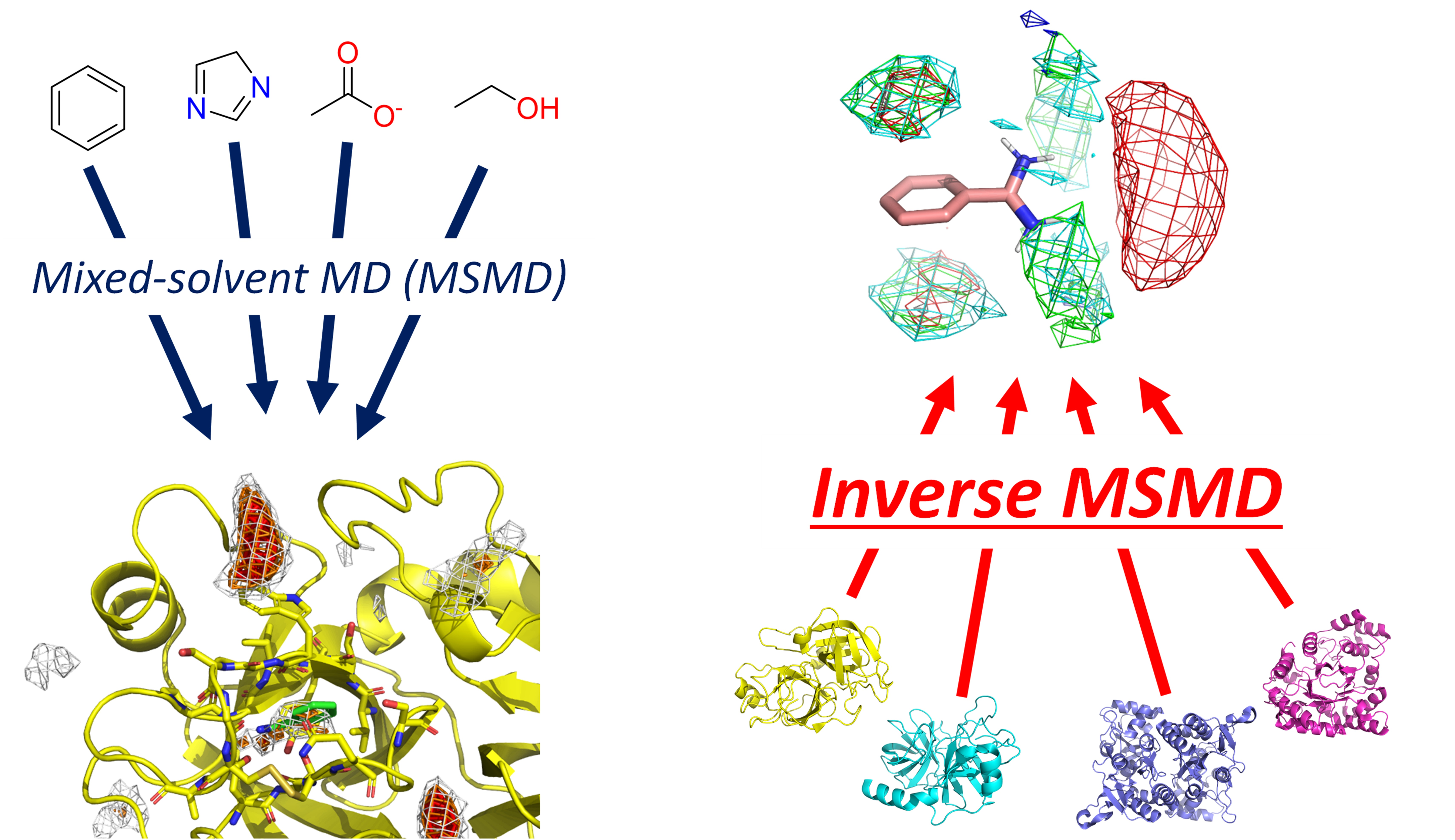
さらに現在、推定したプロファイルを再びタンパク質表面上に適用することで、実際のリガンド結合の強弱を予測する定量的インバース共溶媒MD法の開発にも取り組んでいます。これにより、通常の共溶媒MDと同じことを、標的タンパク質とのMD計算を行うことなく実現することができると期待しています。
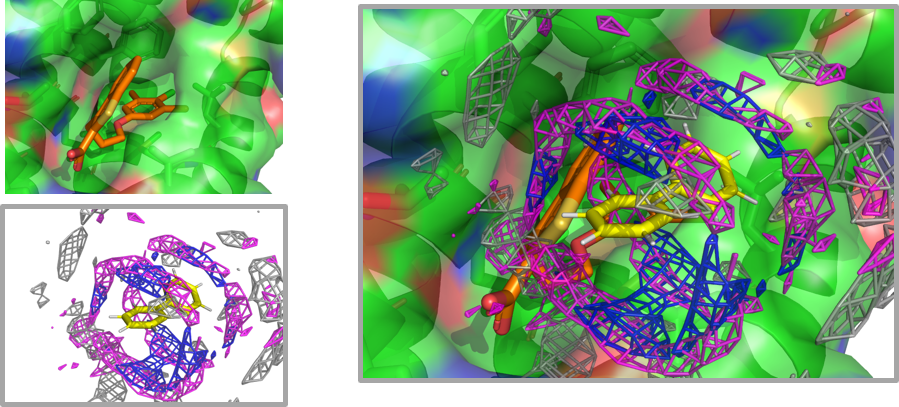
- Keisuke Yanagisawa, Ryunosuke Yoshino, Genki Kudo, Takatsugu Hirokawa. “Inverse Mixed-Solvent Molecular Dynamics for Visualization of the Residue Interaction Profile of Molecular Probes”, International Journal of Molecular Sciences, 23(9): 4749, 2022/4. DOI: 10.3390/ijms23094749
アミノ酸を共溶媒としたMSMD (AAp-MSMD)
MSMDの共溶媒としてアミノ酸分子を用いることで、タンパク質が他のタンパク質やペプチドと相互作用する際の特徴を解析できる手法を開発しました。AAp-MSMD では、20種類のアミノ酸それぞれを小さな共溶媒分子としたMSMDを実行します。この結果得られるアミノ酸残基ごとの「好みの場所」情報は、タンパク質間相互作用 (PPI) やタンパク-ペプチドの結合を予測するのに有用です。なぜなら、タンパク質表面上で特定のアミノ酸が好んで結合する場所が連なれば、そこが数アミノ酸からなるペプチドの結合サイトである可能性があるからです。また、タンパク質上のある残基を変異させた際に、周囲のアミノ酸プローブ分子の付きやすさがどう変化するかを比較することで、点突然変異が結合親和性に与える影響を定性的に評価することも可能です。
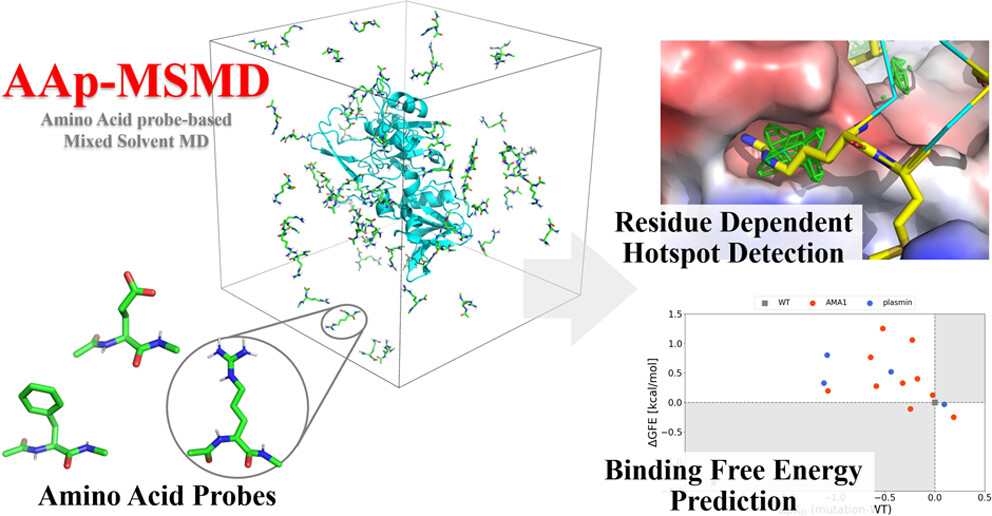
- Genki Kudo, Keisuke Yanagisawa, Ryunosuke Yoshino, Takatsugu Hirokawa. “AAp-MSMD: Amino Acid Preference Mapping on Protein–Protein Interaction Surfaces Using Mixed-Solvent Molecular Dynamics”, Journal of Chemical Information and Modeling, 63(24): 7768-7777, 2023/12. DOI: 10.1021/acs.jcim.3c01677
共溶媒分子セットの構築 (EXPRORER)
MSMD法では、研究の目的や対象となるタンパク質によって様々な種類の共溶媒分子が用いられてきましたが、明確な標準や指針は存在しません。そこで、創薬に用いられる多種多様な化合物群を網羅的にカバーできる共溶媒分子セットの構築を試みました。薬剤分子からフラグメントを100種類以上切り出し、大量のMSMDシミュレーションを実施し、シミュレーション結果同士の類似度を算出、クラスタリングを行うことで、「創薬化合物に典型的に含まれる部分構造」を広くカバーする共溶媒セットを提案しました。
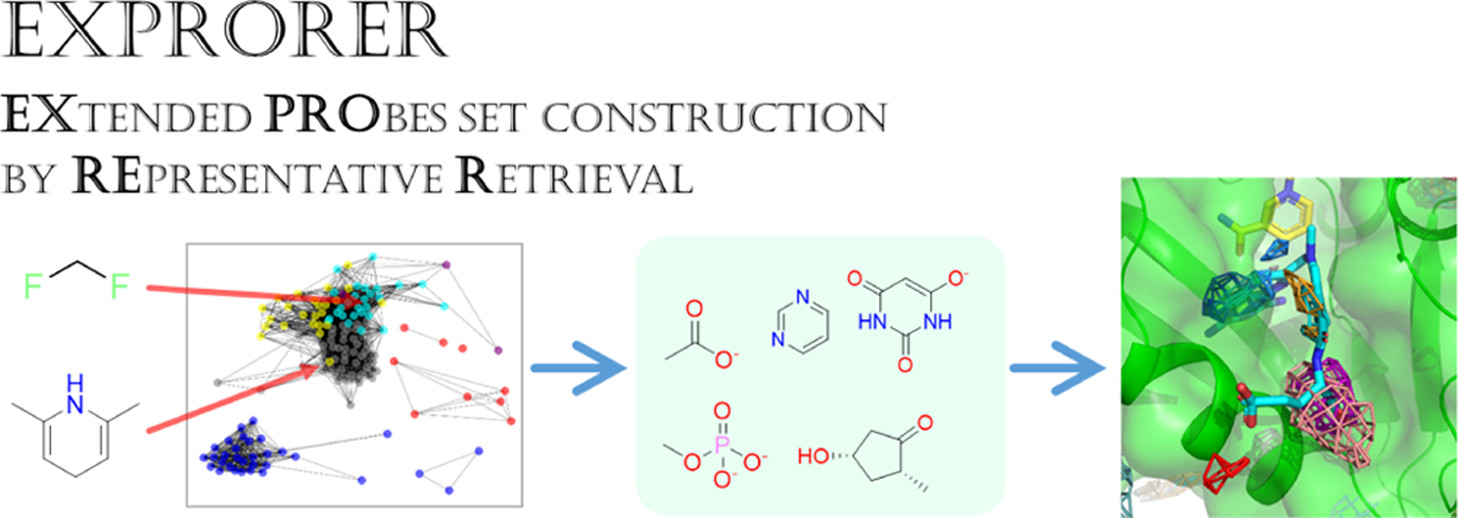
- Keisuke Yanagisawa, Yoshitaka Moriwaki, Tohru Terada, Kentaro Shimizu. “EXPRORER: Rational Cosolvent Set Construction Method for Cosolvent Molecular Dynamics Using Large-Scale Computation”, Journal of Chemical Information and Modeling, 61(6): 2744-2753, 2021/6. DOI: 10.1021/acs.jcim.1c00134
化合物フラグメントに着目した構造ベースバーチャルスクリーニング手法の開発 (2014/4-)
構造ベースバーチャルスクリーニング (Structure-based virtual screening, SBVS) とは、薬剤開発の標的タンパク質の3次元構造を用いて、大量の化合物群から有望な化合物を選抜する処理のことです。この手法は広く注目されていますが、計算時間・精度いずれの面からも性能改善の余地が大きく残されています。
我々は、化合物の部分構造であるフラグメントに着目して研究を行っています。2800万件余りの化合物は、僅か26万件程度のフラグメントで表現できることが知られており、化合物間のフラグメント共通性を利用した計算の高速化を目指しています。
タンパク質化合物ドッキング計算ツール REstretto の開発
多数の化合物を対象とするドッキング計算を効率化するため、フラグメントの再利用に基づくドッキング手法としてREstrettoというツールを開発しました。REstrettoは、一度ドッキングしたフラグメントの情報を保存し、同じフラグメント構造を含む別の化合物のドッキングで再利用することで計算の重複を避けます。これにより、ライブラリー中に存在する類似構造の化合物群をまとめて高速に処理できます。実験的な評価では、約1万件規模の化合物集合に対し、従来広く用いられているAutoDock Vinaと同程度の計算時間とドッキング精度を達成しています。REstrettoはオープンソースソフトウェアとして公開しています。
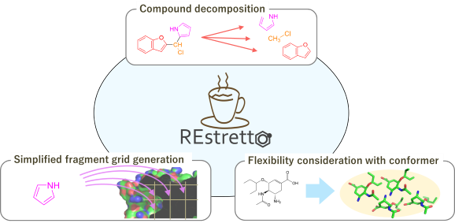
- Keisuke Yanagisawa, Rikuto Kubota, Yasushi Yoshikawa, Masahito Ohue, Yutaka Akiyama. “Effective Protein–Ligand Docking Strategy via Fragment Reuse and a Proof-of-Concept Implementation”, ACS Omega, 7(34): 30265-30274, 2022/8. DOI: 10.1021/acsomega.2c03470
フラグメント分割に基づく高速な化合物プレスクリーニング手法の開発
創薬の初期段階であるバーチャルスクリーニングでは、時に数億を超えるような極めて多数の化合物を評価することも想定されます。しかし、これらを一つひとつドッキング評価するのは計算量的に非現実的であり、化合物の分子量等の物性からその数を削減することが一般的です。しかし、このフィルタリングは標的タンパク質に結合する化合物を除外してしまうことにもつながります。我々は、タンパク質の3次元構造情報を用いつつ高速に化合物をスコア評価できるプリスクリーニング法として、Spresso を開発しました。
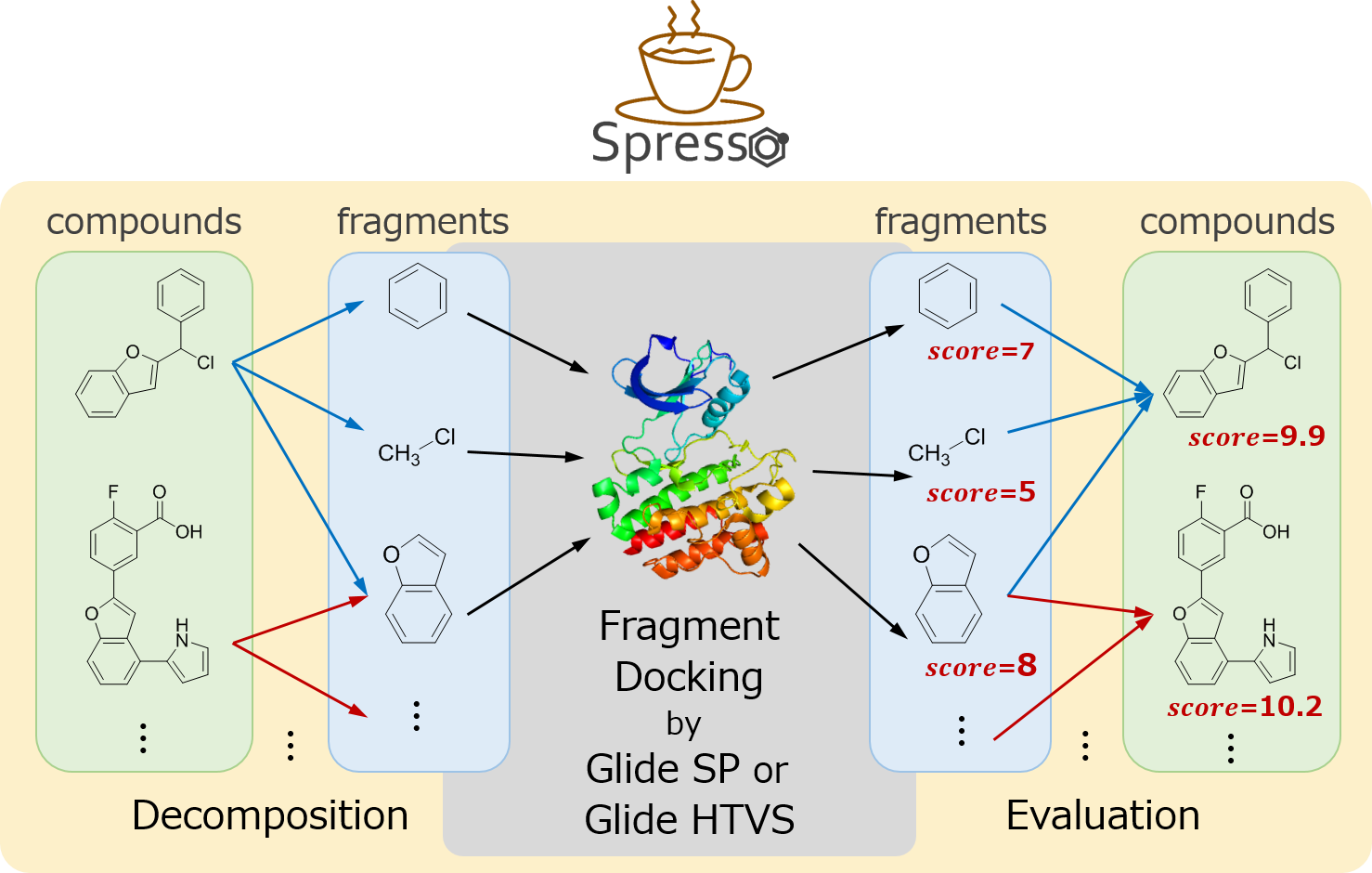
- Keisuke Yanagisawa, Shunta Komine, Shogo D. Suzuki, Masahito Ohue, Takashi Ishida, Yutaka Akiyama: “Spresso: An ultrafast compound pre-screening method based on compound decomposition”, Bioinformatics, 33: 3836-3843, 2017/12 [open access]
- Keisuke Yanagisawa, Shunta Komine, Shogo D. Suzuki, Masahito Ohue, Takashi Ishida, Yutaka Akiyama: “ESPRESSO: An ultrafast compound pre-screening method based on compound decomposition”, The 27th International Conference on Genome Informatics (GIW 2016), 7 pages, 2016/10
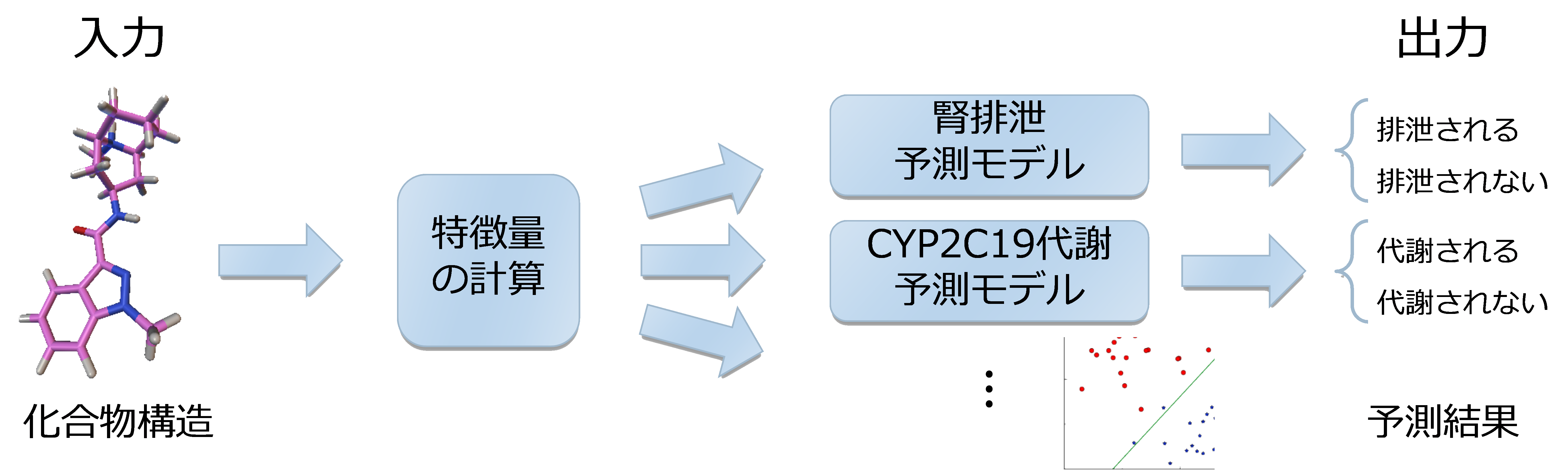
半教師付き学習を用いた薬物クリアランス経路予測 (2013/04-2015/08)
薬物化合物の分子量 (MW)、分配係数 (logD)、血漿中タンパク質非結合率 (fup)などを計算し、これらの値を利用してヒト体内のどのような代謝・排泄経路(クリアランス経路)を通過するかを予測します。
この予測問題は「ラベル付け(クリアランス実験)のコストが非常に高く、ラベル付けされていない化合物構造は大量に存在している」という性質を持っています。 したがって、一般によく用いられる教師付き学習ではなく、ラベル付けされていないデータも利用することの出来る半教師付き学習が予測に適していると考えられます。
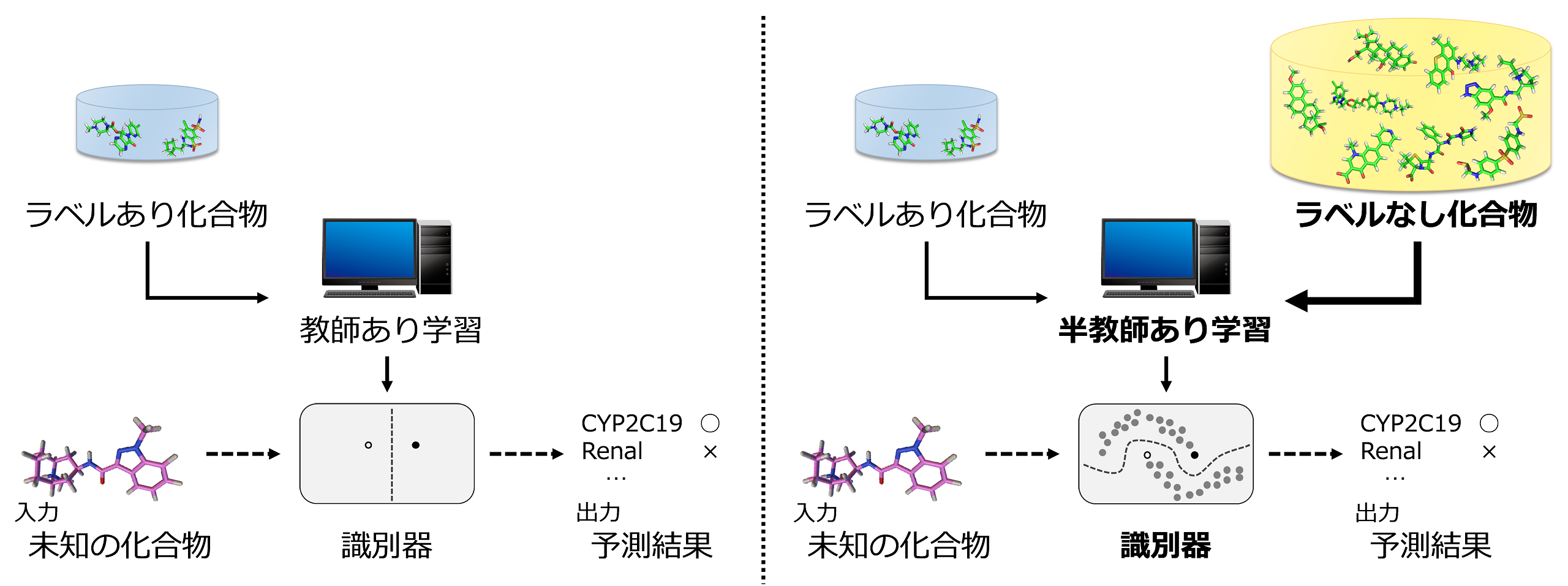
- Keisuke Yanagisawa, Takashi Ishida, Yutaka Akiyama: “Drug clearance pathway prediction based on semi-supervised learning”, IPSJ Transactions on Bioinformatics, 8: 21-27, 2015/08 [open access]