
Docking Calculations Using Quantum Annealer (2023/04-)
A quantum annealer is a type of quantum computer that is expected to rapidly search for local optimal solutions to specific forms of combinatorial optimization problems. We aim to apply this quantum annealer to protein-ligand docking calculations (a method for predicting the binding mode and binding affinity of candidate compounds based on protein three-dimensional structures). To achieve this, we have converted docking calculations into a format solvable by quantum annealer and formulated them as QUBO (quadratic unconstrained binary optimization) problems. Specifically, we divide ligand molecules into multiple fragments (substructures) and represent the placement of each fragment with binary variables, thereby expressing the docking search space as QUBO.
- Keisuke Yanagisawa, Takuya Fujie, Kazuki Takabatake, Yutaka Akiyama. “QUBO Problem Formulation of Fragment-Based Protein–Ligand Flexible Docking”, Entropy, 26(5): 397, 2024/4. DOI: 10.3390/e26050397
Mixed-Solvent Molecular Dynamics (MSMD) (2019/04-)
Mixed-solvent molecular dynamics (MSMD) is a molecular dynamics (MD) method that uses proteins as solutes and water molecules along with co-solvent molecules as solvents. Based on information about where and to what extent co-solvent molecules exist on protein surfaces, MSMD is used for hotspot detection and binding affinity evaluation, as well as for discovering cryptic binding sites (hidden binding sites) by utilizing protein structural changes induced by co-solvent molecules.
We have made various proposals regarding this MSMD method.
Constructing residue interaction profile of probes (Inverse MSMD)
In conventional co-solvent MD, we map where co-solvent molecules are likely to exist on protein surfaces (existence probability distribution). In contrast, inverse MSMD performs estimation of “interaction profiles from the co-solvent perspective,” which determines what types of protein residues are likely to appear around co-solvent molecules.
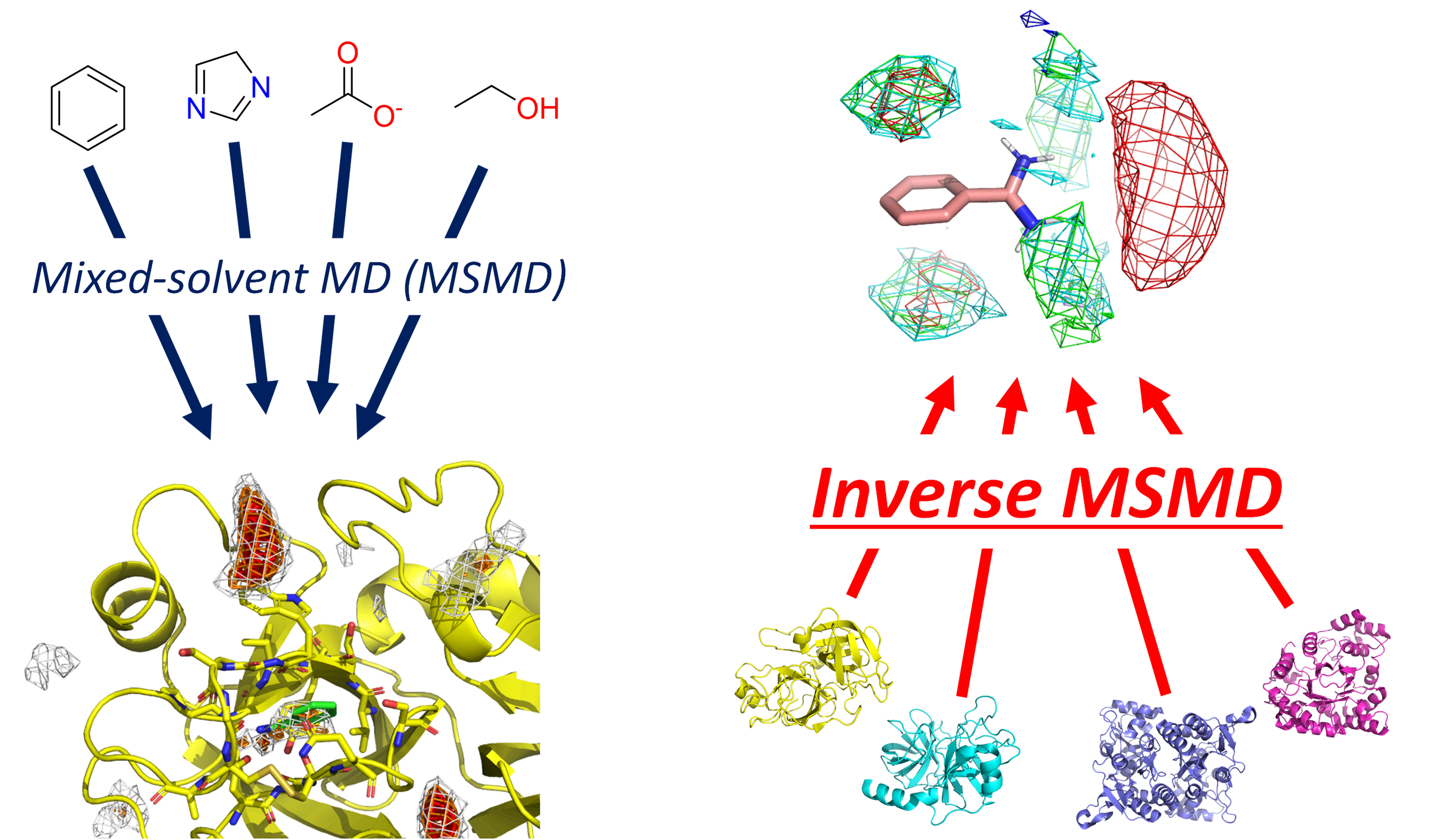
Furthermore, we are currently developing a quantitative inverse co-solvent MD method that predicts the strength of actual ligand binding by applying the estimated profiles back to protein surfaces. This is expected to achieve the same results as conventional co-solvent MD without performing MD calculations with target proteins.
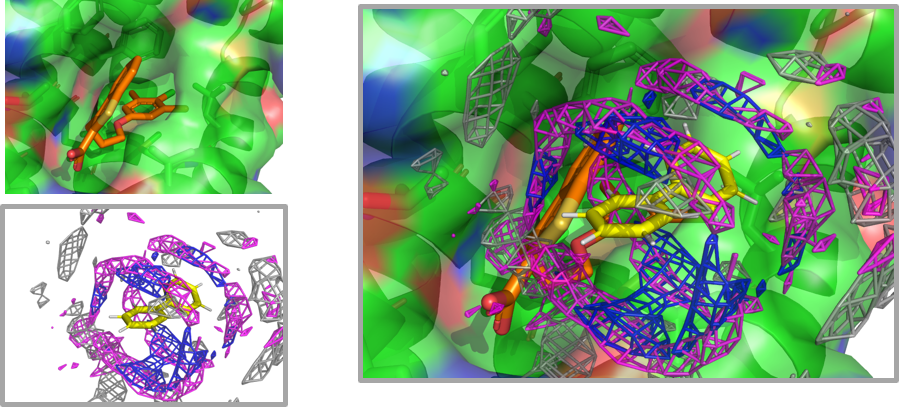
- Keisuke Yanagisawa, Ryunosuke Yoshino, Genki Kudo, Takatsugu Hirokawa. “Inverse Mixed-Solvent Molecular Dynamics for Visualization of the Residue Interaction Profile of Molecular Probes”, International Journal of Molecular Sciences, 23(9): 4749, 2022/4. DOI: 10.3390/ijms23094749
MSMD Using Amino Acids as Co-solvents (AAp-MSMD)
We developed a method that can analyze the characteristics of protein interactions with other proteins or peptides by using amino acid molecules as co-solvents in MSMD. AAp-MSMD performs MSMD using each of the 20 amino acids as small co-solvent molecules. The resulting “preferred location” information for each amino acid residue is useful for predicting protein-protein interactions (PPI) and protein-peptide binding. This is because if locations where specific amino acids preferentially bind on protein surfaces are connected, there is a possibility that this represents a binding site for peptides consisting of several amino acids. It is also possible to qualitatively evaluate the impact of point mutations on binding affinity by comparing how the attachment tendency of surrounding amino acid probe molecules changes when a certain residue on a protein is mutated.
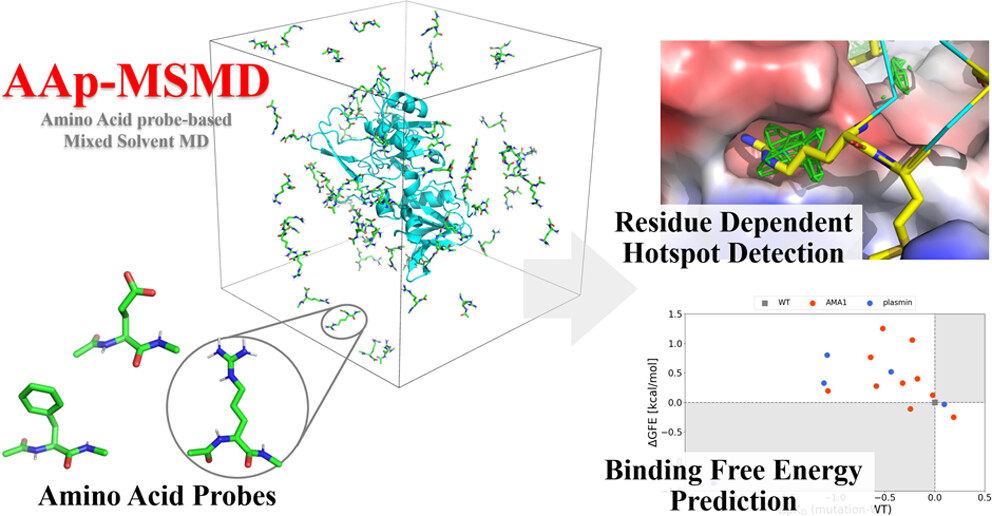
- Genki Kudo, Keisuke Yanagisawa, Ryunosuke Yoshino, Takatsugu Hirokawa. “AAp-MSMD: Amino Acid Preference Mapping on Protein–Protein Interaction Surfaces Using Mixed-Solvent Molecular Dynamics”, Journal of Chemical Information and Modeling, 63(24): 7768-7777, 2023/12. DOI: 10.1021/acs.jcim.3c01677
Construction of Co-solvent Molecule Sets (EXPRORER)
In MSMD methods, various types of co-solvent molecules have been used depending on research purposes and target proteins, but no clear standards or guidelines exist. Therefore, we attempted to construct a co-solvent molecule set that can comprehensively cover the diverse range of compounds used in drug discovery. We extracted over 100 types of fragments from drug molecules, performed large-scale MSMD simulations, calculated similarities between simulation results, and performed clustering to propose a co-solvent set that broadly covers “substructures typically contained in drug discovery compounds”.
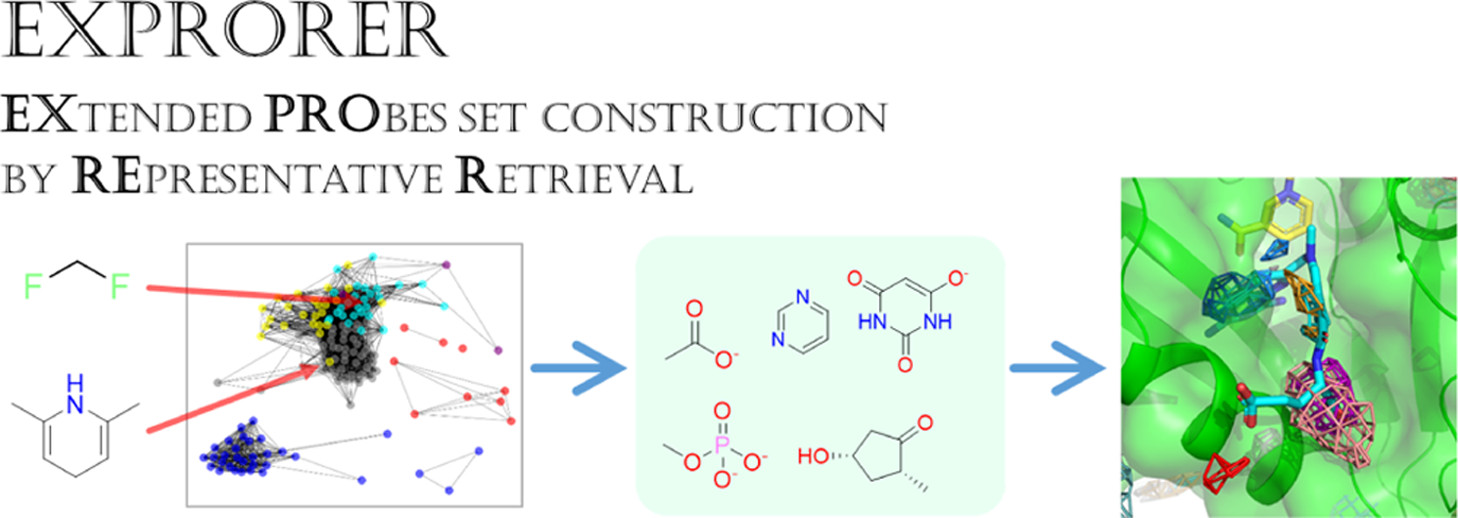
- Keisuke Yanagisawa, Yoshitaka Moriwaki, Tohru Terada, Kentaro Shimizu. “EXPRORER: Rational Cosolvent Set Construction Method for Cosolvent Molecular Dynamics Using Large-Scale Computation”, Journal of Chemical Information and Modeling, 61(6): 2744-2753, 2021/6. DOI: 10.1021/acs.jcim.1c00134
Development of Structure-Based Virtual Screening Methods Focusing on Compound Fragments (2014/4-)
Structure-based virtual screening (SBVS) refers to the process of selecting promising compounds from large compound libraries using the three-dimensional structure of target proteins in drug development. While this method has attracted widespread attention, there remains significant room for performance improvement in terms of both computational time and accuracy.
We focus our research on fragments, which are substructures of compounds. It is known that over 28 million compounds can be represented by only about 260,000 fragments, and we aim to accelerate calculations by utilizing fragment commonality between compounds.
Development of Protein-Compound Docking Calculation Tool REstretto
To improve the efficiency of docking calculations targeting numerous compounds, we developed a tool called REstretto as a docking method based on fragment reuse. REstretto saves information from previously docked fragments and reuses it in docking calculations for other compounds containing the same fragment structures, thereby avoiding computational redundancy. This enables high-speed processing of groups of compounds with similar structures existing in libraries. Experimental evaluation shows that for compound sets of approximately 10,000 compounds, REstretto achieves computational time and docking accuracy comparable to the widely used AutoDock Vina. REstretto is publicly available as open source software.
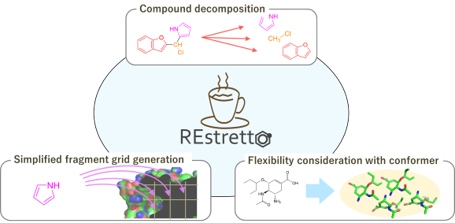
- Keisuke Yanagisawa, Rikuto Kubota, Yasushi Yoshikawa, Masahito Ohue, Yutaka Akiyama. “Effective Protein–Ligand Docking Strategy via Fragment Reuse and a Proof-of-Concept Implementation”, ACS Omega, 7(34): 30265-30274, 2022/8. DOI: 10.1021/acsomega.2c03470
Development of a Fast Compound Pre-screening Methods Based on Compound Decomposition
In virtual screening, which is the initial stage of drug discovery, it is sometimes necessary to evaluate extremely large numbers of compounds, potentially exceeding hundreds of millions. However, performing docking evaluation for each compound individually is computationally unrealistic, so it is common to reduce their number based on compound properties such as molecular weight. However, this filtering can also lead to the exclusion of compounds that bind to target proteins. We developed Spresso as a pre-screening method that can rapidly score compounds while using protein three-dimensional structural information.
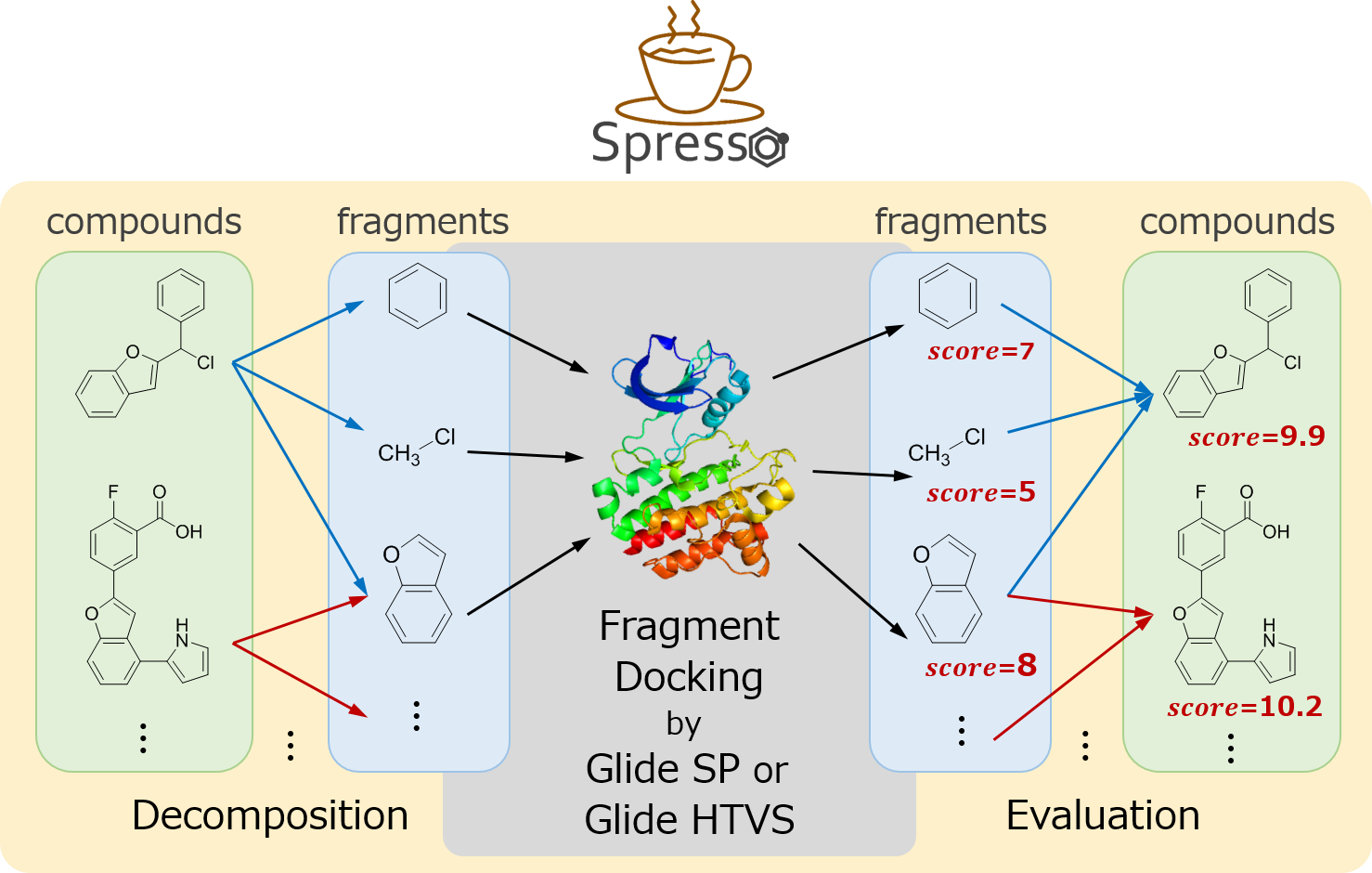
- Keisuke Yanagisawa, Shunta Komine, Shogo D Suzuki, Masahito Ohue, Takashi Ishida, Yutaka Akiyama. “Spresso: an ultrafast compound pre-screening method based on compound decomposition”, Bioinformatics, 33(23): 3836-3843, 2017/12. DOI: 10.1093/bioinformatics/btx178
- Keisuke Yanagisawa, Shunta Komine, Shogo D. Suzuki, Masahito Ohue, Takashi Ishida, Yutaka Akiyama. “ESPRESSO: An ultrafast compound pre-screening method based on compound decomposition”, The 27th International Conference on Genome Informatics (GIW 2016), 2016/10/4.
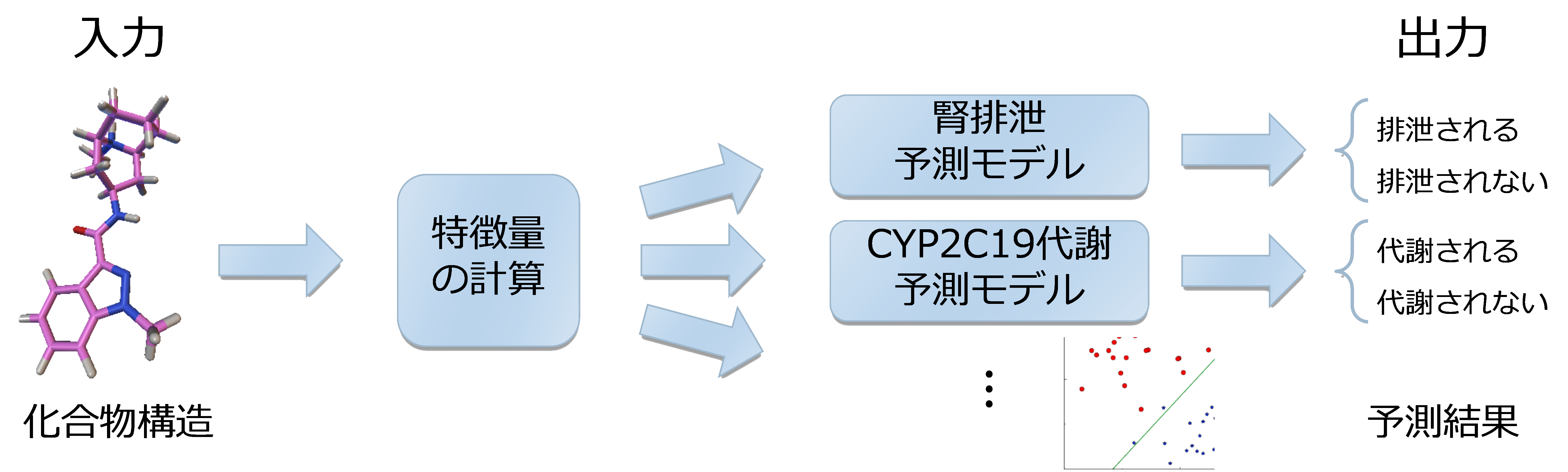
Drug Clearance Pathway Prediction Using Semi-Supervised Learning (2013/04-2015/08)
We calculate molecular weight (MW), partition coefficient (logD), plasma protein unbound fraction (fup), and other properties of drug compounds, and use these values to predict which metabolic and excretion pathways (clearance pathways) they will pass through in the human body.
This prediction problem has the characteristic that “the cost of labeling (clearance experiments) is very high, and there are large amounts of unlabeled compound structures available.” Therefore, semi-supervised learning, which can utilize unlabeled data, is considered more suitable for prediction than commonly used supervised learning.
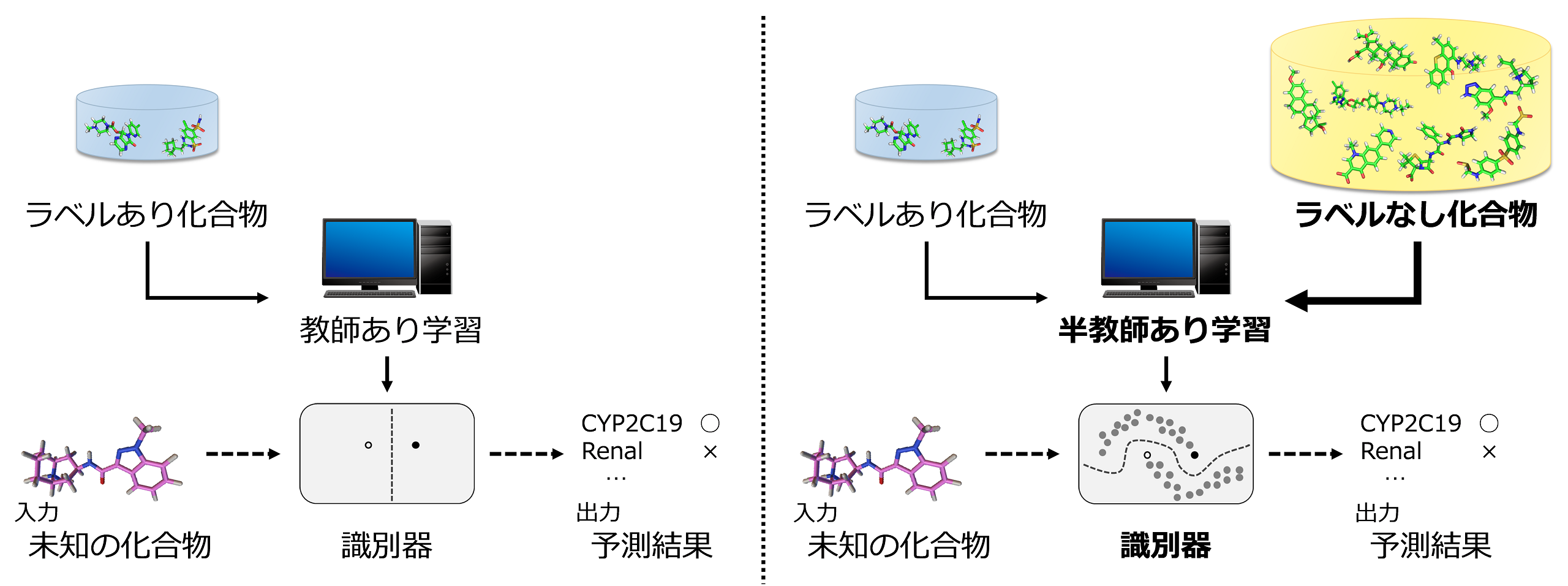
- Keisuke Yanagisawa, Takashi Ishida, Yutaka Akiyama: “Drug clearance pathway prediction based on semi-supervised learning”, IPSJ Transactions on Bioinformatics, 8: 21-27, 2015/08 [open access]